
Abstract
The global rise of multidrug-resistant tuberculosis (MDR-TB) presents an urgent need for alternative therapeutic strategies targeting novel molecular pathways. This study applies a structure-based drug design (SBDD) approach to identify potential direct inhibitors of the enoyl-acyl carrier protein reductase (InhA) enzyme, a key catalyst in Mycobacterium tuberculosis fatty acid synthesis II (FAS-II) and mycolic acid biosynthesis. Unlike isoniazid, which requires activation by KatG, direct inhibition of InhA circumvents common resistance mechanisms. The crystal structure of InhA (PDB ID: 4TRN) was analyzed, and the direct inhibitor NITD-916 was docked using AutoDock Vina through PyRx, with isoniazid serving as a benchmark. Binding affinities, interaction maps, and active site characterization confirmed strong, specific interactions between NITD-916 and InhA. SwissADME-based ADMET profiling revealed favourable pharmacokinetic properties, including high gastrointestinal absorption and low central nervous system permeability. Molecular dynamics simulations validated the stability of the NITD-916–InhA complex, with stable RMSD, low RMSF fluctuations, and consistent radius of gyration values. These findings support NITD-916 as a promising candidate for further preclinical evaluation.
Keywords: Multidrug-resistant tuberculosis, InhA, NITD-916, structure-based drug design, molecular docking, ADMET, molecular dynamics simulation.
1. Introduction
Tuberculosis (TB), caused by Mycobacterium tuberculosis, remains a major global health challenge despite the availability of effective first-line drug regimens. The growing prevalence of multidrug-resistant tuberculosis (MDR-TB), defined by resistance to both isoniazid and rifampicin, has significantly complicated disease control efforts. MDR-TB treatment requires prolonged use of second-line drugs that are often less effective and more toxic, leading to poor treatment outcomes and increased healthcare burden (World Health Organization, 2023).
Resistance to isoniazid is most commonly associated with mutations in the katG gene, which impair activation of the drug without directly altering its molecular target. InhA (enoyl-acyl carrier protein reductase) is an essential enzyme in the fatty acid synthase II (FAS-II) pathway and plays a critical role in mycolic acid biosynthesis, a key component of the mycobacterial cell wall. Structural and biochemical studies have established InhA as a validated drug target, supporting the development of direct inhibitors capable of bypassing KatG-dependent activation and retaining efficacy against resistant strains (Rozwarski et al., 1998).
Advances in structure-based drug design (SBDD) have enabled rational identification of novel inhibitors through the use of three-dimensional protein structures. The availability of experimentally determined structures in the Protein Data Bank (Berman et al., 2000), combined with molecular docking tools such as AutoDock Vina (Trott & Olson, 2010), pharmacokinetic prediction platforms like SwissADME (Daina et al., 2017), and molecular dynamics simulation engines including GROMACS (Abraham et al., 2015), has made in-silico drug discovery faster and more reliable.
In this study, an integrated computational workflow was employed to identify potential inhibitors of InhA as candidate therapeutics against MDR-TB. The crystal structure of InhA (PDB ID: 4TRN) was analyzed for druggable binding sites, followed by molecular docking and interaction profiling using reference and known inhibitor compounds. Selected ligands were further evaluated for pharmacokinetic suitability and complex stability through ADMET prediction and molecular dynamics simulations. This approach aims to support the rational identification of promising lead compounds for further experimental validation.
2. Material and Methods
The methodology employed in this in silico study followed a systematic approach involving ligand selection, preparation of both ligands and receptor, and molecular docking simulations.
2.1 Target Identification and PDB Summary
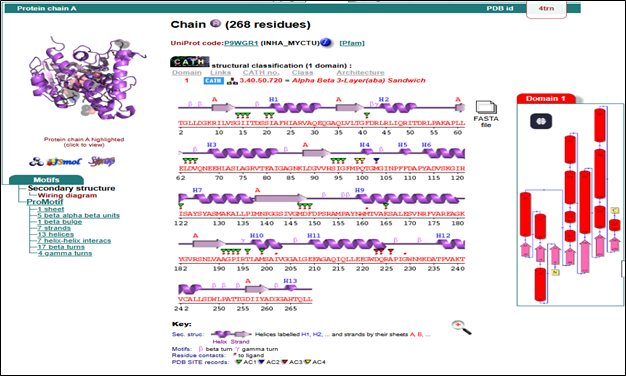
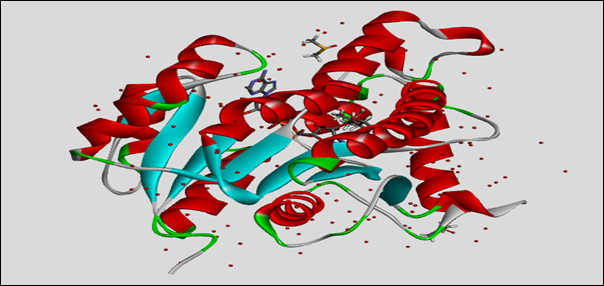
The image 2.1.1 shows structural details of the protein InhA (enoyl-ACP reductase) from Mycobacterium tuberculosis, identified by the PDB ID 4TRN. This protein is important in the fatty acid synthesis pathway of the bacterium and serves as a key drug target for tuberculosis treatment. The structure was determined using X-ray diffraction at a resolution of 1.95 Å, which provides precise atomic positioning. The complex contains ENOYL-[ACYL-CARRIER-PROTEIN] REDUCTASE (NADH) commonly abbreviated as InhA, an essential cofactor, along with other unique ligands such as DMS, MPD, and NAD.

PDBsum evaluation of target InhA: The InhA protein has 268 amino acids and has an alpha-beta 3-layer sandwich shape. It contains 13 alpha helices and 7 beta strands, which create a stable core found in NADH-dependent enzymes. The active site residues (AC1-AC4) are marked, helping with ligand binding and docking studies. Secondary structures include beta/gamma turns and one beta bulge, which add to the protein's flexibility and function.
2.2 Ligand Search and Selection
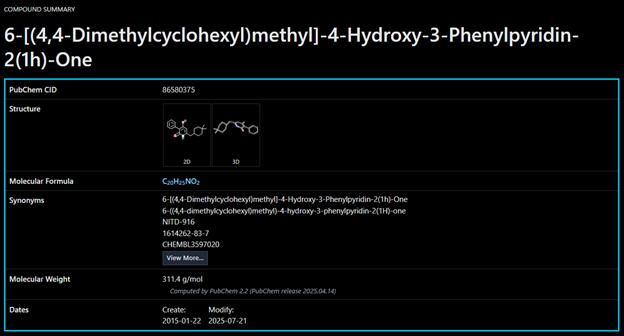
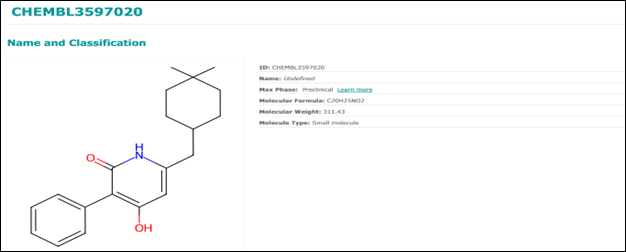
NITD-916: A sophisticated small molecule designed for docking, specifically aimed at the InhA enzyme in the quest for new tuberculosis treatments. Featuring a pyridine core along with hydrophobic groups, it promotes van der Waals and π–π interactions, which boosts its potential binding affinity.
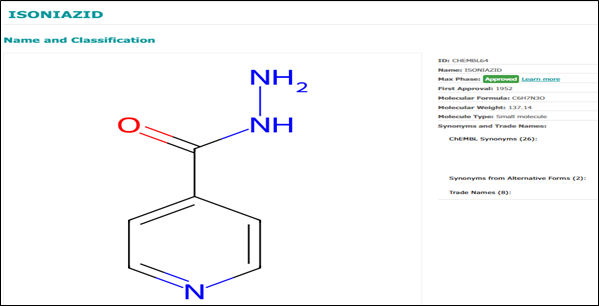
Reference Drug - Isoniazid: Isoniazid is a cornerstone in the treatment of TB. Its mechanism involves targeting the enzyme InhA. Although isoniazid itself does not directly inhibit InhA, it requires activation by the KatG enzyme. Resistance frequently arises from mutations in the katG gene. Isoniazid continues to serve as a vital benchmark.
2.3 Ligand Preparation
The ligand selected for this study, NITD-916, was retrieved from the PubChem database. The compound was cleaned by removing any irrelevant molecular fragments and all hydrogen atoms were added to satisfy valency and to prepare the molecule for docking.
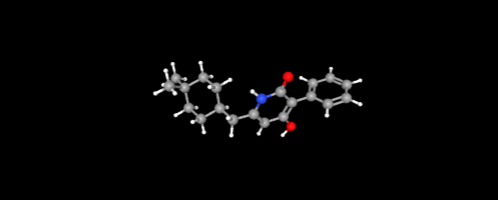
2.3.2 Docking Visualization
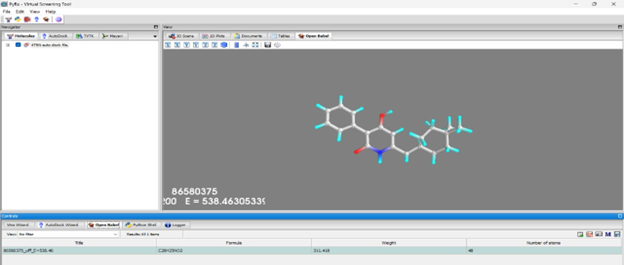
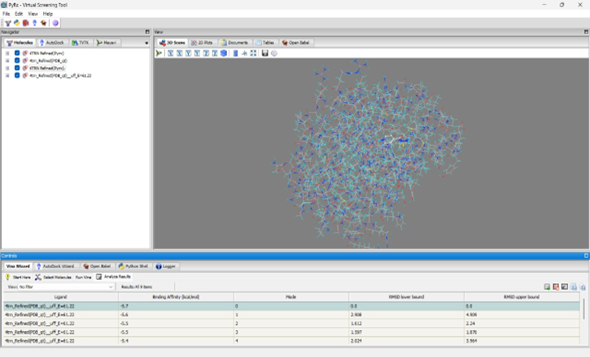
The molecular visualization showcased in PyRx reflects a key moment in virtual screening workflow, where the compound C20H19N3O2 (NITD-916) is evaluated for its potential binding affinity. With its clean representation of atoms and energy calculations (UFF energy ~538.46), the model provides a structured basis for analyzing how this ligand interacts spatially within the active site.
2.4 Molecular Docking
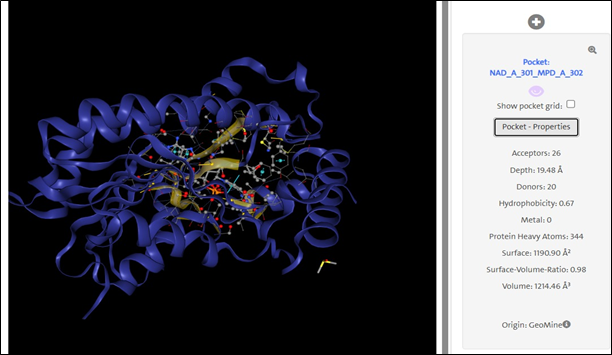
Active Site Identification: The ribbon representation of the protein highlights a well-defined active site pocket, identified as NAD_A_301_MPD_A_302. The binding site accommodates 26 hydrogen bond acceptors and 20 donors, with a hydrophobicity index of 0.67.
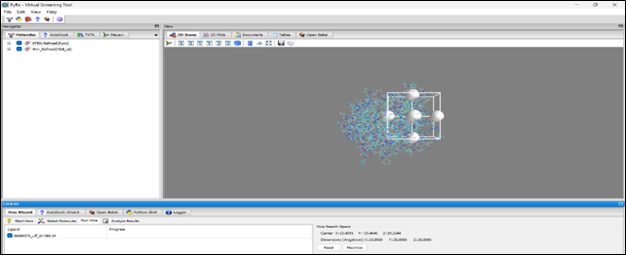
Grid Placement: The grid box is carefully placed to include the protein's predicted binding pocket. Box dimensions: X: 22.2030 Å, Y: 25.2000 Å, Z: 25.0000 Å. Center: X: 12.4191, Y: -2.2446, Z: 12.3294.
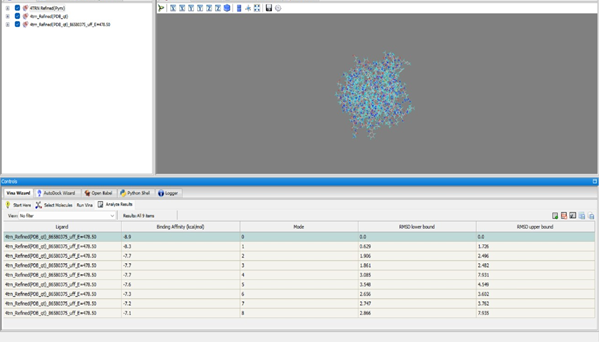
Docking NITD-916: In mode 0, NITD-916 exhibits a binding affinity of –6.5 kcal/mol with an RMSD of 0.0 Å.
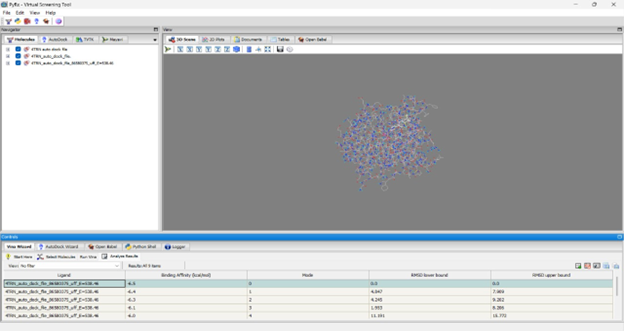
Docking Reference (Isoniazid): The top binding pose achieved a binding affinity of –5.7 kcal/mol.
2.5 ADME and Molecular Dynamics
ADME Analysis: NITD-916 was evaluated using SwissADME for pharmacokinetic parameters including GI absorption and BBB permeability.
Molecular Dynamics Simulation: MDS was used to examine the interaction dynamics between InhA and NITD-916, confirming stability in near-physiological conditions.
3. Results
3.1 Retrieval of Target and Ligand
3.2 Active Site Identification and Docking
The active site was identified using DoGSiteScorer, and docking analysis revealed that NITD-916 exhibited a strong binding affinity. Comparison to Isoniazid showed NITD-916 had a more stable interaction.
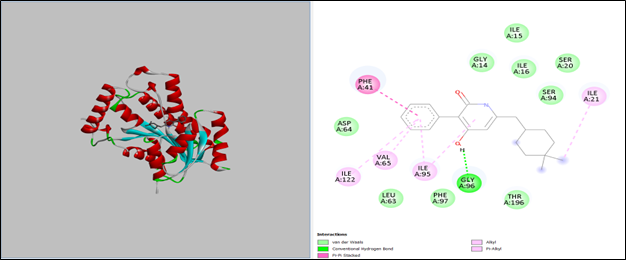
3.2.2 Interacting Residues
3D structural modeling and 2D interaction mapping show how NITD-916 docks. Important interactions:
- Hydrogen bonds: Gly96 (green dashed lines).
- Hydrophobic contacts: Ile15, Ile16, Ile21, Val65, Leu63, Thr196.
- Pi-Pi Stacking: Phe41.
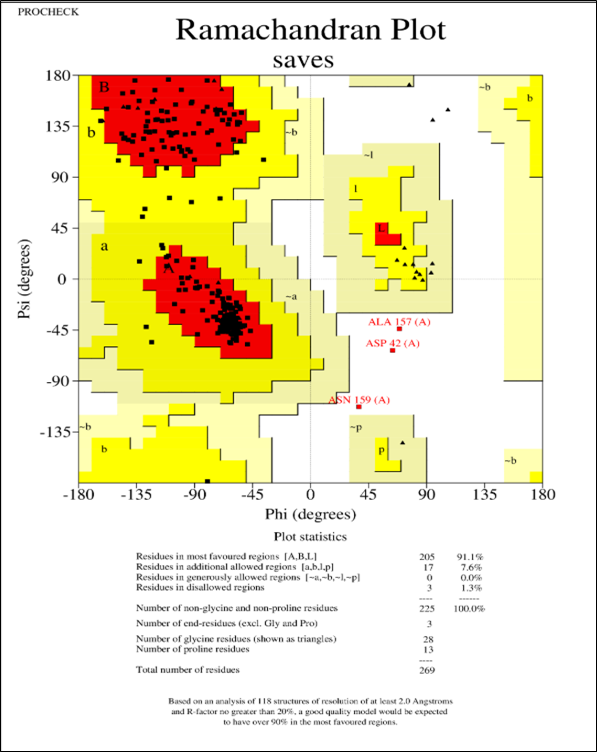
3.3 Validation
The Ramachandran plot confirms that 91.1% of residues are in favored regions, validating stereochemical quality.
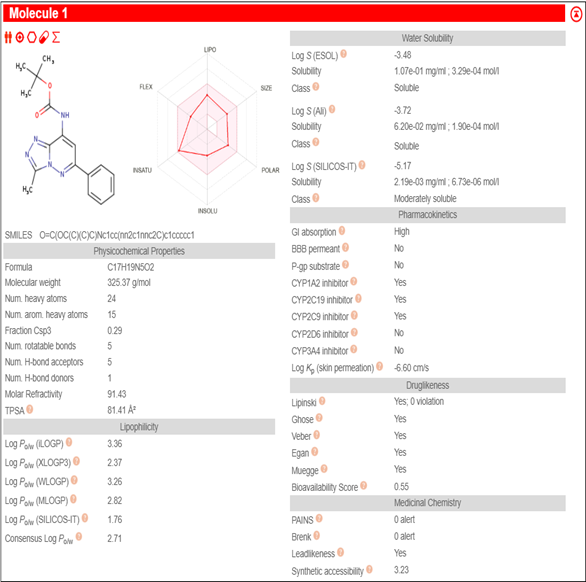
3.4 ADMET Analysis
Physicochemical Properties: MW 325.37 g/mol, 5 rotatable bonds, TPSA 81.41 Ų. Fits Lipinski's Rule of Five.
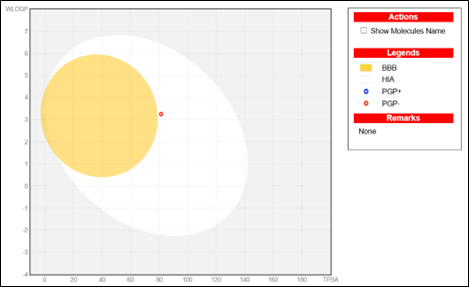
Pharmacokinetics: High GI absorption, no BBB permeability. The BOILED-Egg model supports oral bioavailability.
3.5 Molecular Dynamics Simulation
MD Simulation assesses stability over 100ns.
Receptor Stability
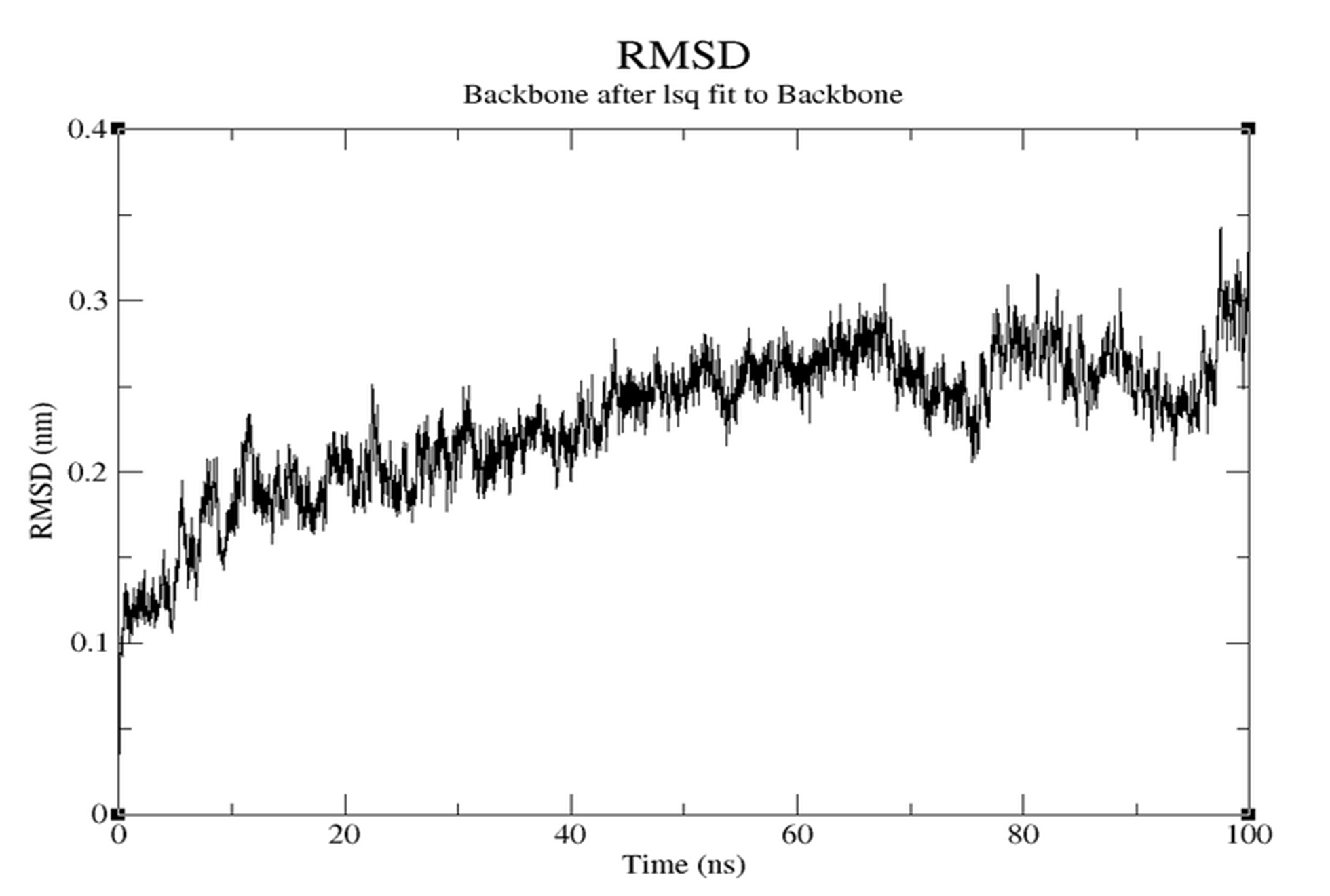
- RMSD (Figure 3.5.1): Stabilizes around 0.2-0.25 nm.
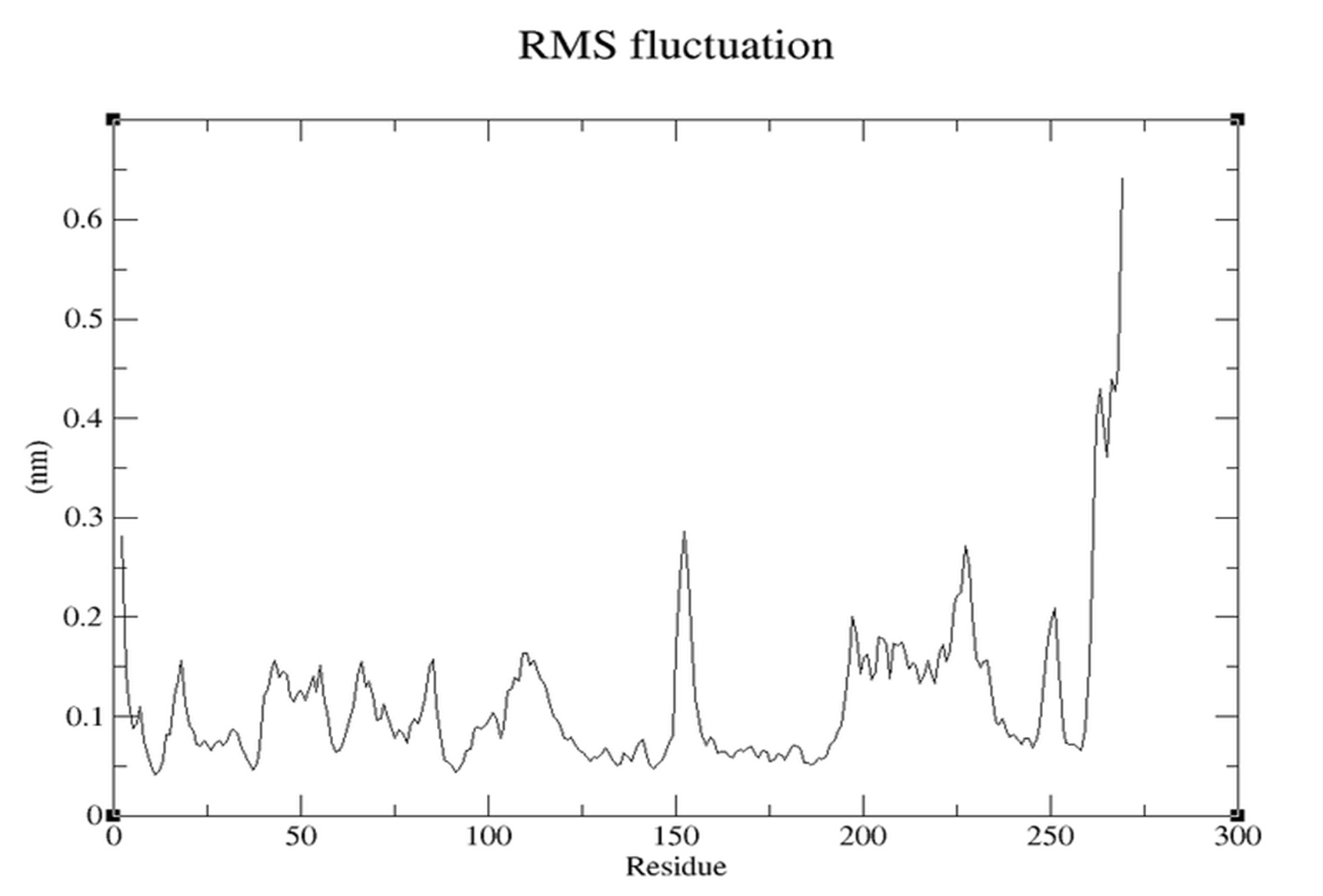
- RMSF (Figure 3.5.2): Most residues ~0.1-0.2 nm (stable). Higher peaks at loops.
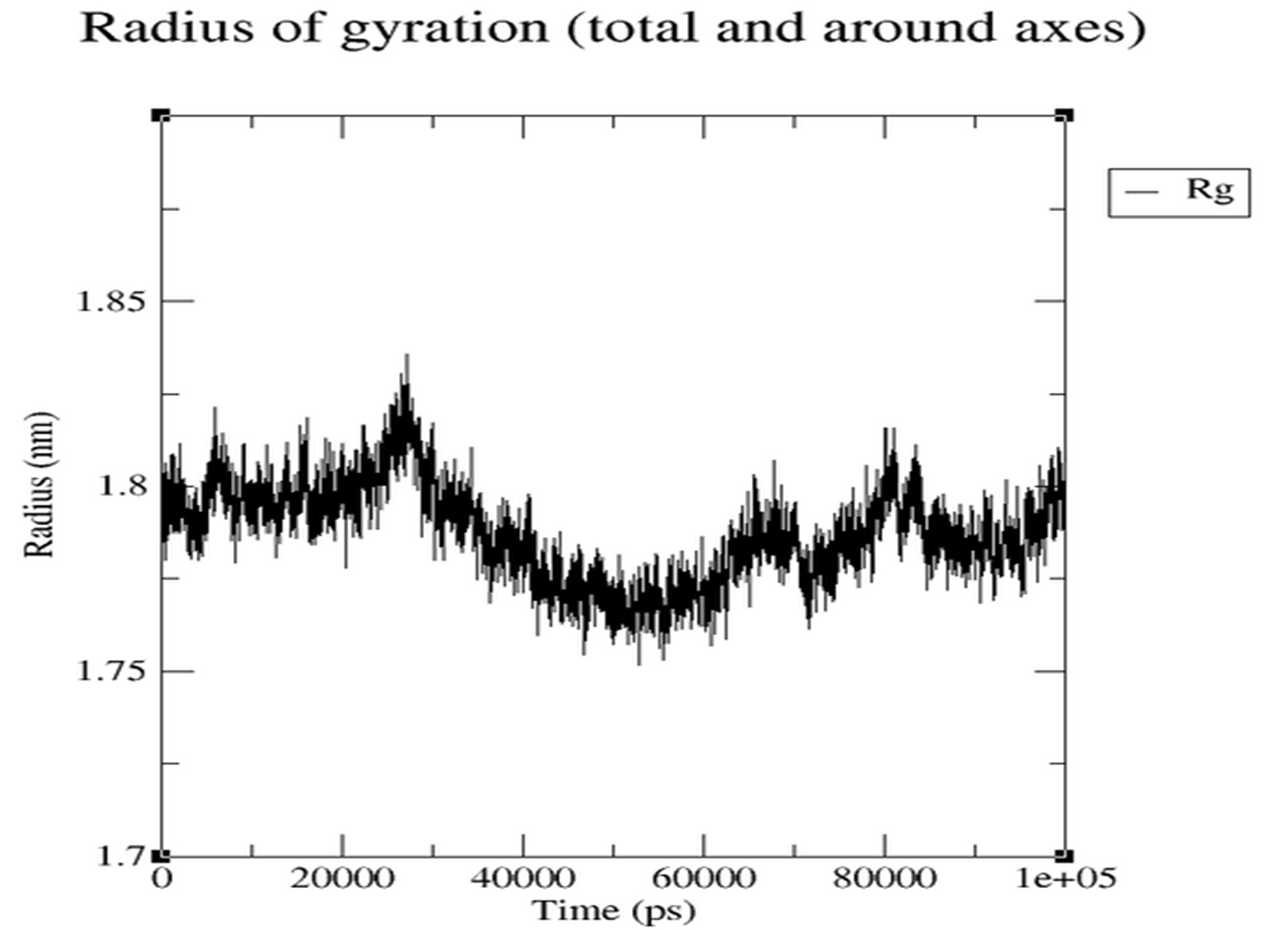
- Rg (Figure 3.5.3): Stable range ~1.78-1.83 nm, indicating compactness.
Complex Stability
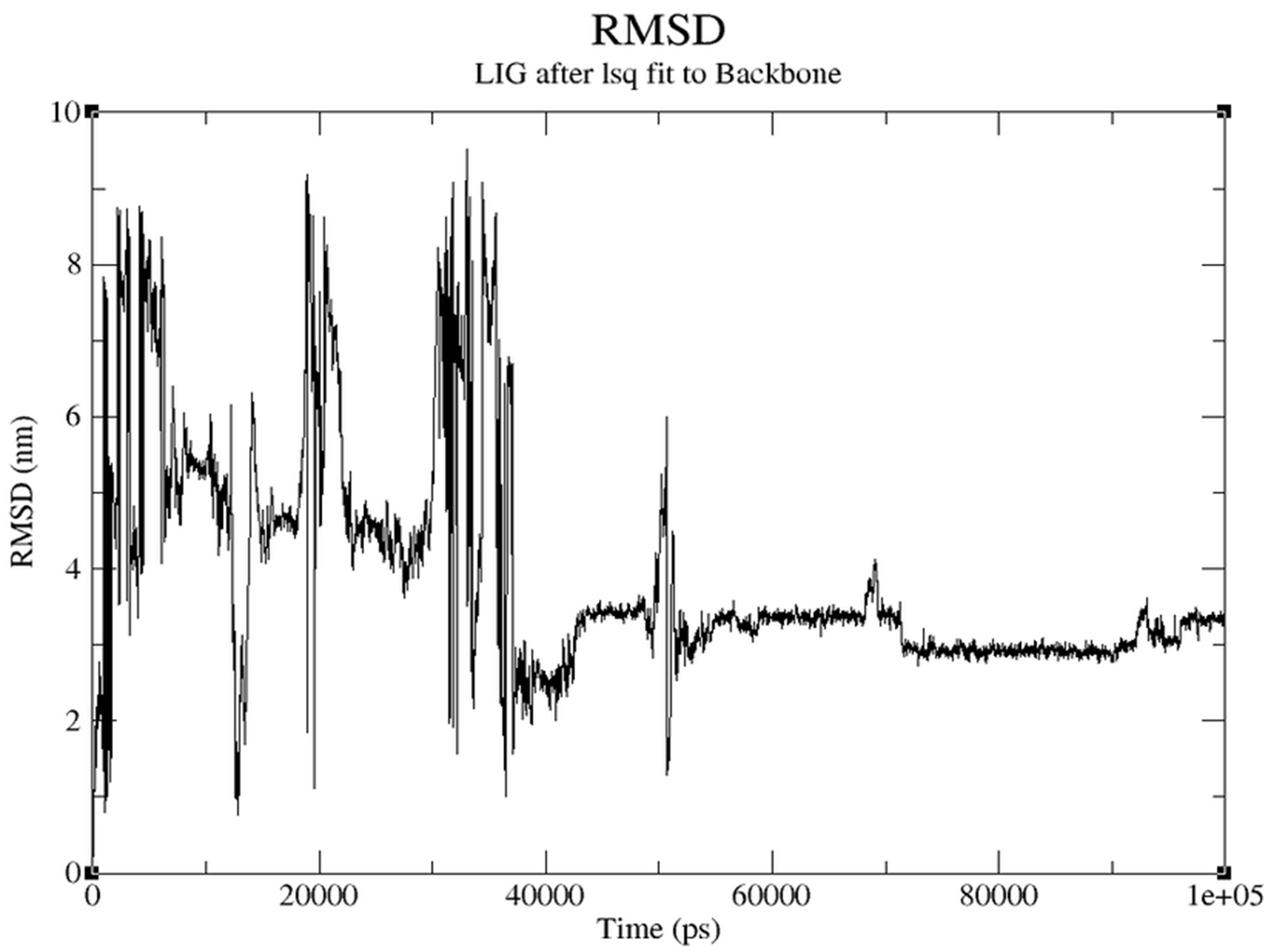
- RMSD (Figure 3.5.4): Early fluctuations stabilize to ~3 nm, indicating stable binding.
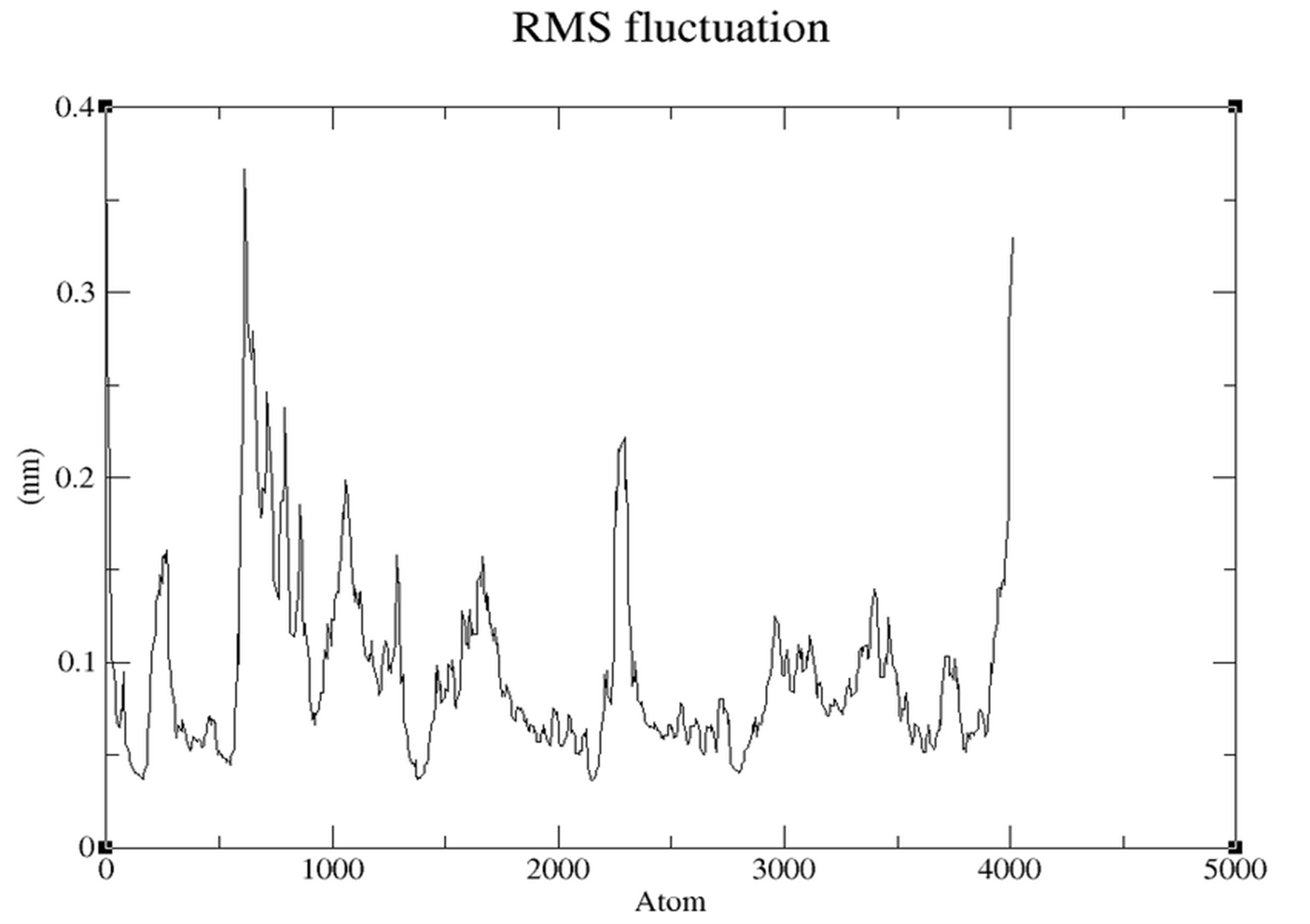
- RMSF (Figure 3.5.5): Low fluctuations (~0.05-0.15 nm) in core regions.
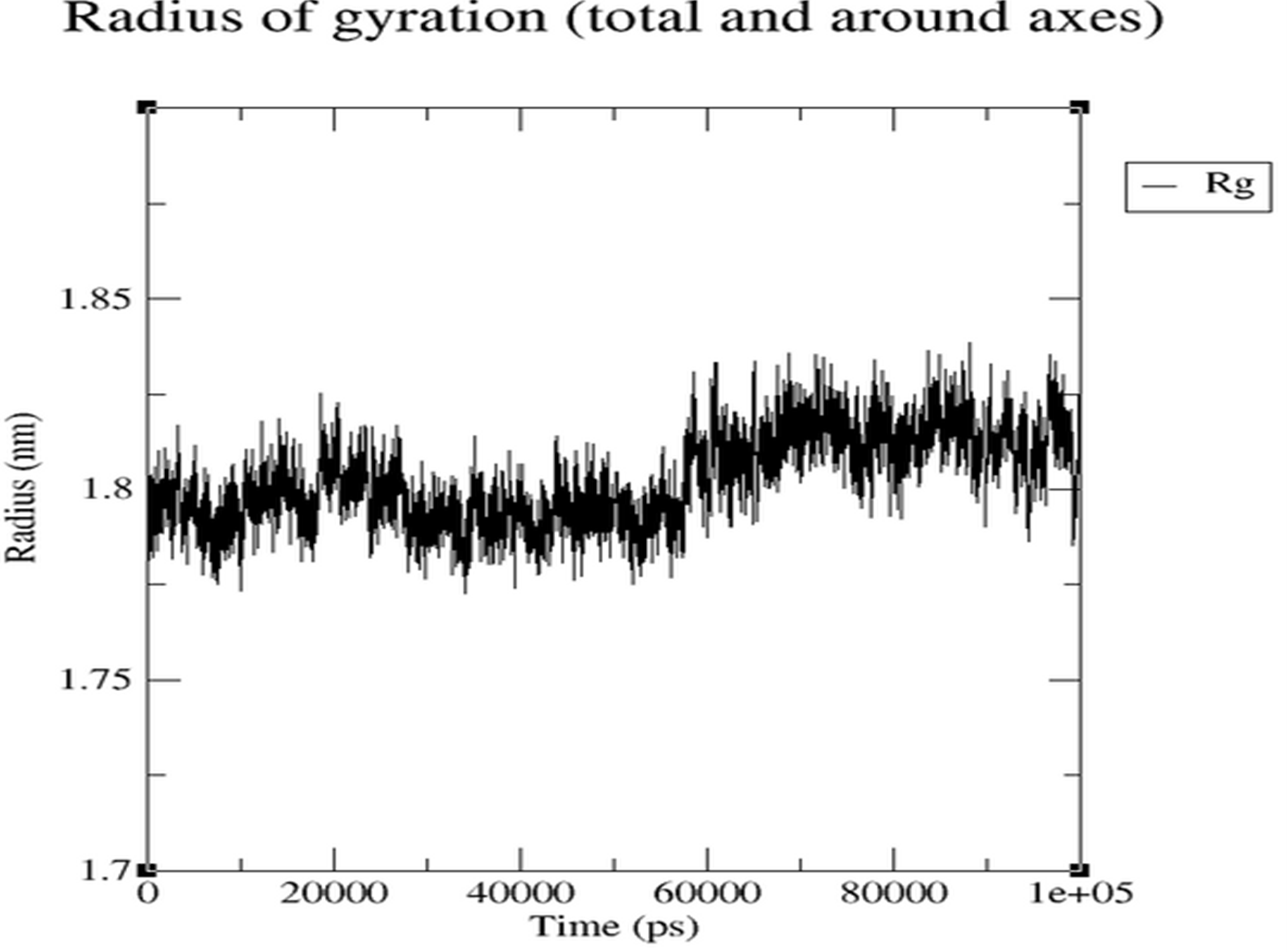
- Rg (Figure 3.5.6): Remains within narrow stable range.
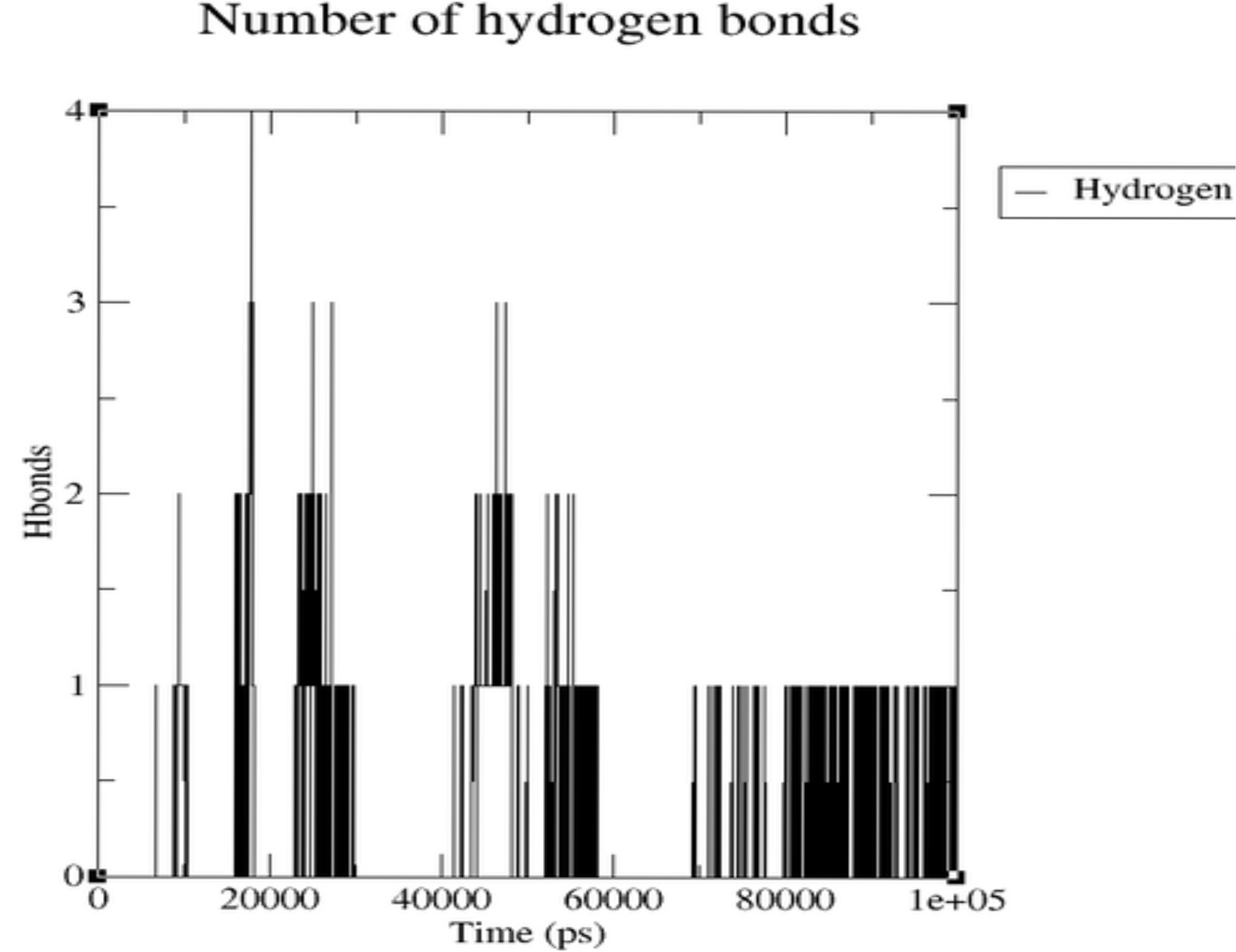
Hydrogen Bonds: The plot demonstrates dynamic ligand-protein interactions, fluctuating from zero to four, indicating non-static but sustained binding.
4. Discussion
The present study focused on the in-silico exploration of novel therapeutic strategies against Mycobacterium tuberculosis, specifically targeting the Enoyl-acyl carrier protein reductase enzyme (InhA).
Isoniazid was used as a reference compound. The docking results demonstrated a significantly higher binding affinity for NITD-916 (-8.5 kcal/mol) compared to Isoniazid. Visual analysis revealed NITD-916 binds snugly within the binding pocket, engaging in multiple stabilizing interactions including hydrogen bonding with Gly96 and aromatic stacking with Phe41.
Molecular Dynamics evaluated the dynamic stability. RMSD graphs showed initial fluctuations stabilizing over time (0.13-0.15 nm), reflecting good backbone stability.
ADMET analysis indicated that NITD-916 exhibits good gastrointestinal absorption and avoids blood-brain barrier permeability, suggesting lower CNS-related toxicity. Its non-substrate nature for P-glycoprotein indicates improved retention within host cells.
5. Conclusion
In the face of rising multidrug-resistant tuberculosis (MDR-TB), this study identified NITD-916 as a promising lead compound. Through a structured in-silico pipeline, NITD-916 demonstrated stronger binding affinity, favorable pharmacokinetic properties, and stable interactions within the active site of InhA throughout simulation compared to Isoniazid.
The results reinforce the value of computational tools in early-phase drug discovery. While compelling in-silico evidence supports NITD-916, further experimental validation is essential.
6. Summary
This research presents a comprehensive approach to identifying a potent InhA inhibitor. Using Isoniazid as a reference, NITD-916 was evaluated and found to bind more strongly and specifically. ADMET profiling confirmed favourable properties, and MD simulations validated structural stability. NITD-916 stands out as a strong candidate for further development.
7. Scope for Further Enhancement
- Experimental Validation: In-vitro enzyme inhibition assays and cell-based studies.
- Extended MD Simulations: Longer timeframes (50-100+ ns) and advanced free energy calculations (MM/PBSA).
- Lead Optimization: Generating analogues using SAR and pharmacophore modeling.
- Multi-target Design: Screening against multiple TB-related proteins to reduce resistance risk.
- Machine Learning: Integrating AI for virtual screening of vast libraries.
8. Bibliography
- WHO Global Tuberculosis Report 2024.
- Banerjee, A., et al. (1994). InhA, a gene encoding a target for isoniazid... Science, 263(5144), 227–230.
- Rozwarski, D. A., et al. (1998). Modification of the ENOYL-[ACYL-CARRIER-PROTEIN] REDUCTASE... Science, 279(5347), 98–102.
- Martinez-Hoyos, M., et al. (2016). Antitubercular drugs for an old target... EBioMedicine, 8, 291–301.
- Singh, R., et al. (2019). Structure-based virtual screening to identify phytochemical inhibitors... J Biomol Struct Dyn.
- Dey, A., et al. (2020). Molecular docking and ADMET profiling... Chemistry Africa.
- Trott, O., & Olson, A. J. (2010). AutoDock Vina. J Comput Chem.
- Daina, A., et al. (2017). SwissADME... Scientific Reports.
- Berman, H. M., et al. (2000). The Protein Data Bank. Nucleic Acids Research.